��������
��������
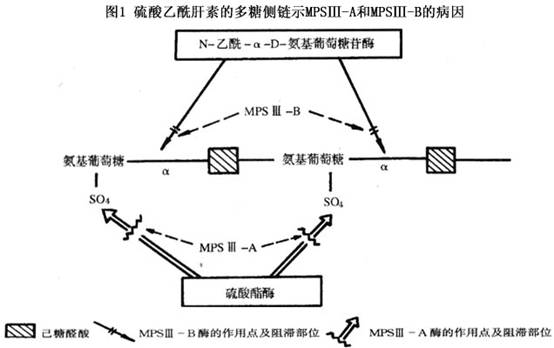
�������ƣ����ݳ���άϸ���������������ɽ�������ΪA��B��C��D���͡�A����������ø����������-N-������øȱ������B��������N-����-��-D-������������ø(ͼ1)����C������������CoA-��-�����ǰ�-N-����ת��øȱ������D��������N-����-��-D-������������-6-������øȱ����������øȱ��ʹ�����������ز������⣬���ڸμ���֯�������������������ų�����Ȼø��ȱ�ݲ�ͬ����4�͵�֢״��ȫ��ͬ���ٴ��ϲ������֡�4��ϸ�������������лȱ�ݣ�ʹ4�Ͳ���ϸ���ڵ����dz������õ����������֡��������������������Ϊ��ͬ�Ͳ��˵ij���άϸ�����������ϸ��ȱ����ø���¡�
�������������ɼ���ϸ���Ϳ��ո�ϸ���Լ�ֱ���Ĥϸ�����д�����γɡ�����������ʵ����Ԫ���ر��Ժ���ʧ������������Ԫ�������䣬�ں�֬�ʶ�������ͣ��Ի�����֬�������ࡣ�����ں�����ϸ���������͡��Ե���Щ��������ٴ�������
����������ԭ��
�羵�����ʾ�в�ͬ���͵�ϸ���ʰ����壻���о��ȿ������ʼ�Сͬ��Բ��ĤС����ݣ�������ˮ��Ĥ�Ͱ���С����ݡ�
��֯��ѧ���֤���ԡ��Ρ�����������������ҪΪ�����������أ�����֯�л�����֬��GM����֬�������ӡ�
�ٴ�����
�ٴ����֣�������
���������DZ�֢������������������1�����������������������������
��������һ����4��7����֣�10��ʱ�Ѻ����ء���
�������������Լ��ص�ͬʱ���ɳ��ֽ�������֢״�����硢�˶����ࡢ��������̱֫��ȫ����������������Ϊ�ȣ���Ϊ��֢��ͻ����֢״��
���ĸı������������������1/4�IJ��˱��ְ�С������������沿�����������������˱���ͷ�����ݳ�ª��������¡��������
�������ؽڽ�ֱ����������צ״�������ı���ж�ԳɹDz�ȫ�Ͷ��Ǻ����ܡ�������Щ�ı����ϱ�������һ���̶ȵ������ԡ��Ρ�
Ƣ�״�Ϊ��ȵ��жȣ���Ĥ���ǡ����������ۡ�
Ԥ��
Ԥ����
��ԭ�ۻ�����һ���ͯ�Ŵ�����ԭ��л���ҵļ�������Ҫ�Ի�����֯��ԭ�ۻ�������ֽ�����Ϊ�ص㣬����Ϊ��ԭ�ϳɴ�л�ϰ�����ʹ������֯��ԭ�����١�
��ԭ�ۻ�������Ϊһ�ּ���������һ�鼲����Ŀǰȷ��������12�֡��ٴ�����
��Ѫ��Ϊ���������漰����������ҪΪ�Ρ��������������Ŵ���ʽ��Ϊ��Ⱦɫ�������Ŵ������Ա���죬���ڶ�ͯ�ڷ��������ֲ��������˺��鲻�ٷ�չ������ά��һ�㽡��ˮƽ��
������Ҫ������ȱ���ֽ���ԭ��ijЩø����������-6-����ø����-1��4������߰ø��������Ǽ�ø�������ữ��ø�ȡ�
����ߵĸ�ĸΪ����飬����������Ԥ����������Ҫ���ڡ�һ��������ԭ�ۻ�֢���Է���
��Ѫ��Ϊ������ʳ������ͣ�����֬�����������������������Ѫ��������ߣ��˷�̼�����Ʒ������ж���Ƥ�ʼ��ء��������ء��ȸ�Ѫ���صȿɰ�������
��Ѫ����